Convivere con la sclerosi multipla
Testo di Rosanna Novara Topino |
Malattia neurologica del giovane adulto, la sclerosi non è mortale ma potenzialmente invalidante. In Italia si contano 3.400 nuovi casi all’anno. Oggi la diagnosi precoce e la ricerca scientifica stanno dando risultati importanti.
La sclerosi multipla (Sm), detta anche sclerosi a placche, è una malattia in aumento soprattutto nella popolazione femminile. Si tratta di una patologia infiammatoria su base autornimmune del sistema nervoso centrale (Snc), caratterizzata dalla comparsa di lesioni rotondeggianti e ben delimitate (placche) in molteplici aree dell’encefalo e del midollo spinale. Tali lesioni possono essere rosee e mollicce oppure grigiastre e dure. Le loro dimensioni variano da pochi millimetri a diversi centimetri e sono caratterizzate dalla scomparsa progressiva della mielina presente nella sostanza bianca e dalla proliferazione delle cellule della neuroglia. Queste ultime sono cellule connettivali, che formano l’impalcatura di sostegno dei neuroni e provvedono al loro mantenimento. Nel Snc coesistono la sostanza bianca e quella grigia.
Sostanza bianca e grigia
La sostanza grigia è costituita dai corpi cellulari dei neuroni, dal primo tratto del loro assone (fibra nervosa lunga, per il trasporto degli impulsi nervosi in uscita dal neurone; insiemi di assoni formano i nervi) e dai dendriti (fibre nervose brevi e ramificate per gli impulsi in ingresso nel neurone). È localizzata nella corteccia cerebrale e nella parte centrale del midollo allungato e spinale.
Si è visto che la degenerazione di alcuni assoni comincia presto, all’inizio della malattia, comportando la morte del neurone corrispondente e conseguente deficit funzionale, ovvero alterazione delle prestazioni sensitivo-motorie e, a lungo termine, compromissione cognitiva e comportamentale. All’inizio della malattia, tuttavia, gli assoni degenerati non sono numerosi, per cui i deficit funzionali sono dovuti soprattutto alla flogosi (infiammazione). Quando quest’ultima si risolve, il paziente recupera completamente o in parte le sue funzioni.
Evoluzione della Sm
 Come colpisce questa patologia? La Sm evolve da una fase acuta infiammatoria iniziale ad una fase cronica, quindi, una volta insorta, sarà presente per sempre nella vita del paziente.
Come colpisce questa patologia? La Sm evolve da una fase acuta infiammatoria iniziale ad una fase cronica, quindi, una volta insorta, sarà presente per sempre nella vita del paziente.
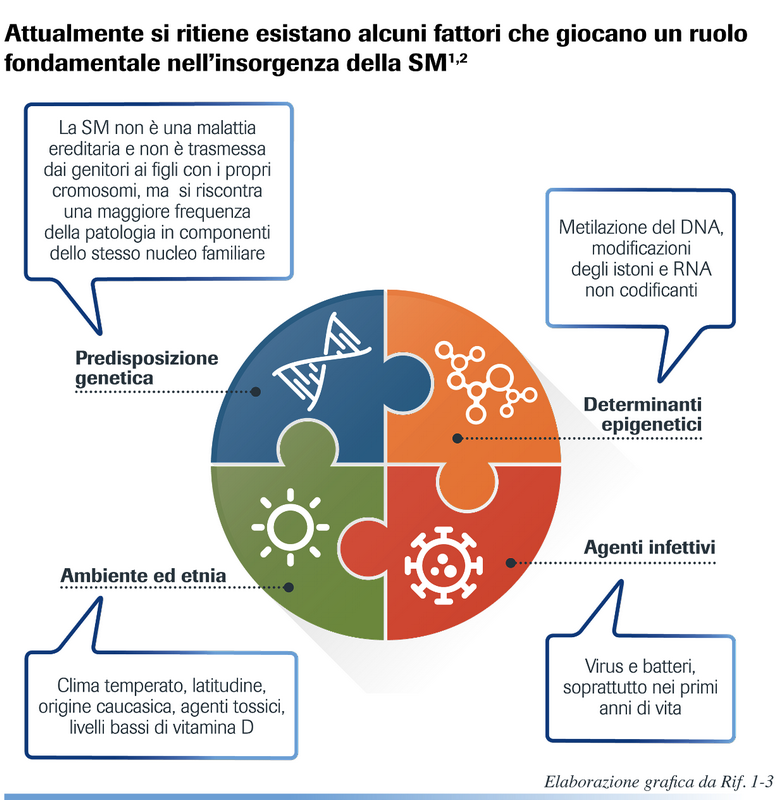
Questa malattia è caratterizzata dalla disseminazione spaziale e temporale delle placche, cioè le aree di demielinizzazione della sostanza bianca compaiono in zone diverse del Snc e in tempi diversi. Il danno mielinico, che caratterizza la Sm è conseguente ad un’anomala attivazione del sistema immunitario sia cellulare che umorale indotta da fattori genetici e ambientali. Questi ultimi sono probabilmente rappresentati da virus come quello del morbillo (verso il quale sono stati trovati anticorpi nel liquor dei pazienti in quantità superiore rispetto ai controlli), ma finora nessun virus è stato correlato con certezza alla Sm. Sicuramente i fattori genetici hanno un peso importante nella predisposizione ad ammalarsi, come è dimostrato dalla concordanza di casi tra i gemelli monozigotici (20-30%), che scende al 4% tra i gemelli dizigotici. I geni coinvolti sembrano essere numerosi. Tra questi ci sono quelli del maggior complesso di istocompatibilità (DR2, DQW1), quelli che codificano per gli interferoni, per le catene leggere dei linfociti T, per il Tnf (Tumoral Necrosis Factor). Recentemente è stata identificata una mutazione genetica del tipo «nonsenso» (in presenza della quale, anziché esserci la codifica di un aminoacido, viene dato il segnale di stop alla sintesi proteica), direttamente collegata alla Sm, localizzata sul gene NR1H3; tale mutazione causa la perdita di funzione del prodotto genico, la proteina LXRA, che controlla l’espressione di diversi geni coinvolti nell’omeostasi dei lipidi (costituenti della mielina, oltre alle proteine), nei processi infiammatori e immunitari. È chiaro quindi che, perché si sviluppi la malattia, è necessaria una predisposizione genetica, su cui devono intervenire dei fattori esogeni (grafico di pagina 63). Gli studi condotti sulle migrazioni di popolazioni hanno evidenziato che c’è un fattore ambientale (forse un virus), che predispone alla Sm e che agisce prima della pubertà (14-15 anni). Un soggetto emigrato prima di questa età acquisisce la prevalenza del paese in cui si è recato, mentre se emigra dopo mantiene la prevalenza del paese d’origine. Tra i virus maggiormente sospettati come parti in causa per la Sm ci sono i Coronavirus, i Papovavirus, come già detto il virus del morbillo, gli Herpesvirus, il Simian V5 e i Retrovirus. Altri fattori di rischio esogeni sono la carenza di vitamina D, per la sua regolazione del sistema immunitario e l’esposizione al fumo di sigaretta.
Giovani e donne
Si stima che al mondo vi siano 2,5-3 milioni di malati di Sm. In Europa essi sarebbero circa 600.000 e in Italia 110.000. Nel nostro paese ci sono oltre 3.400 nuovi casi all’anno. Si prevede che in media una persona su 1.000 avrà la Sm nel prossimo futuro. Il rischio di ammalarsi è maggiore per le donne rispetto agli uomini (2:1). Si sospetta che questo divario sia dovuto a fattori neuroendocrini, che influenzano a più livelli il sistema immunitario. Quest’ultimo, attivato in modo anomalo, colpisce la mielina dell’organismo considerandola come non-self, ovvero estranea (autornimmunità). Il coinvolgimento dei fattori ormonali nella prevalenza della Sm nelle donne è indirettamente dimostrato dal fatto che essa è più frequente in giovani donne in età fertile e dagli effetti della gravidanza e del parto sulle ricadute (queste ultime diminuiscono durante la gravidanza, in particolare nel 2°-3° trimestre e aumentano nel puerperio). Rischiano di ammalarsi 30-50/1.000 persone, se hanno uno dei genitori ammalato di Sm, mentre se sono ammalati entrambi i genitori, il rischio sale a 120/1.000.
L’età di esordio di questa patologia è tra i 15-50 anni e colpisce prevalentemente le persone di 20-30 anni, mentre si rileva raramente sotto i 12 anni e sopra i 55. Per frequenza, la Sm è la seconda malattia neurologica del giovane adulto, dopo i traumi al Snc derivati dagli incidenti stradali e la prima di tipo infiammatorio cronico.
Tipologie di Sm
 Nel 1869 il neurologo Jean Martin Charcot definì i sintomi clinici della malattia e i primi criteri diagnostici, raggruppati nella triade che da lui prende il nome: nistagmo, tremore intenzionale e parola scandita.
Nel 1869 il neurologo Jean Martin Charcot definì i sintomi clinici della malattia e i primi criteri diagnostici, raggruppati nella triade che da lui prende il nome: nistagmo, tremore intenzionale e parola scandita.
Nonostante nel tempo la Sm presenti un’evoluzione diversa da persona a persona, è possibile classificare le varie forme di decorso clinico in quattro gruppi principali: a ricadute e remissioni (SmRR), secondariamente progressiva (SmSP), primariamente progressiva (SmPP) e progressiva con ricadute.
La forma di Sm più diffusa all’esordio è la recidivante-remittente o SmRR (circa 85% tra le 4 forme principali), la quale presenta attacchi ben definiti (pousses), che si risolvono completamente o parzialmente dopo diverse ore o giorni, seguiti da periodi di benessere (remissioni) di durata variabile. Tra i sintomi più frequenti ci sono i problemi alla vista (diplopia), la spasticità o rigidità, le funzioni sfinteriche e viscerali alterate, la minzione imperiosa o urgenza urinaria. Tra un attacco e l’altro, la malattia può rimanere inattiva per mesi o anni.
Il passaggio alla forma secondariamente progressiva (SmSP) avviene dopo circa 10 anni. In questa seconda fase, la malattia cambia gradualmente dal processo infiammatorio tipico della SmRR alla progressione costante, caratterizzata dallo sviluppo di disabilità progressive, che non escludono la sovrapposizione di recidive.
Nella Sm primariamente progressiva (SmPP) non ci sono vere e proprie ricadute. I malati di questa forma di Sm (meno del 10%) presentano fino dall’esordio della malattia dei sintomi, che progrediscono in modo graduale, senza fasi di remissione. Le persone colpite da questa forma hanno più lesioni a livello del midollo spinale che dell’encefalo, tendono ad avere maggiori problemi di deambulazione e il rapporto maschi / femmine colpiti è circa 1:1.
La Sm progressiva con ricadute (SmPR) è una forma poco comune in cui, oltre ad un andamento progressivo fin dall’inizio, sono presenti anche episodi acuti di malattia, con scarso recupero. Passando dalla forma remittente-recidivante a quella secondariamente progressiva, può essere presente questa forma per qualche tempo.
A queste forme principali si aggiungono la Sm benigna, che, al contrario di tutte le altre forme, non peggiora col passare del tempo, esordisce con uno o due episodi acuti, che si risolvono senza lasciare disabilità o quasi e la Sm maligna di Marbourg o Sm fulminante acuta o tumefattiva, una rara forma con decorso tumultuoso ed esito letale nel giro di un anno o due circa. Ultimamente questa forma è risultata sensibile ad alcuni farmaci antitumorali ed al trapianto di cellule staminali.
Alcuni ricercatori ritengono che le forme benigne di Sm siano il 20% di quelle con diagnosi clinica.
Conseguenze della Sm
Tranne la forma maligna di Marbourg, la Sm non è una malattia che porta a morte il paziente, ma purtroppo in molti casi a grave invalidità.
Nonostante la ricerca faccia costantemente nuove scoperte, la causa primaria della malattia resta sconosciuta. Si sa che il sistema immunitario attacca la proteina basica della mielina, considerandola estranea all’organismo, grazie alle sue cellule, che superano la barriera emato-encefalica di difesa e si dirigono nel sistema nervoso centrale, provocando infiammazione e perdita della mielina.
Anche se c’è una predisposizione genetica, tuttavia la Sm non è una malattia ereditaria. È quindi infondato il timore di trasmetterla ai figli (al massimo viene trasmessa la predisposizione ad ammalarsi, che però necessita, come visto, anche di fattori ambientali per dare origine alla malattia). Questo significa che per le donne con Sm, che lo desiderano, non è impossibile intraprendere una gravidanza. È però necessario valutare attentamente la necessità di essere aiutate nell’accudimento dei figli, nel momento in cui dovessero presentarsi ricadute della malattia.
Tra i sintomi della Sm, oltre a quelli già citati precedentemente, ci sono anche i problemi di equilibrio e di coordinazione (compromissione delle funzioni cerebellari), le sensazioni alterate (formicolii, parestesie, dolore), senso di affaticamento, disturbi della sfera sessuale come l’impotenza, disturbi affettivi, ansia, depressione (dapprima psicogena reattiva e successivamente biologica da squilibrio dei sistemi serotoninergici).
Dopo circa 20-30 anni di malattia, compare un decadimento cognitivo nella maggior parte dei pazienti.
Le terapie attuali e future
Le terapie attualmente usate sono sintomatiche per limitare i danni delle ricadute e di tipo immunosoppressivo per allungare il più possibile i tempi di remissione della malattia e rallentarne la progressione. Già da alcuni anni però sono in corso ricerche, che prevedono l’uso di cellule staminali, nel tentativo di stimolare la produzione di nuova mielina, per ridurre il danno assonale.
Per i malati di Sm è fondamentale l’aiuto dei loro cari o comunque di persone che se ne prendano cura non solo per la disabilità a cui vanno incontro, ma per gli stati depressivi in cui possono trovarsi e che possono portare a tentativi di suicidio (il rischio di suicidio tra i malati di Sm è di 1:15, rispetto alla popolazione normale). La depressione può ridurre l’aderenza alla terapia, le performance cognitive e aumentare il senso di affaticamento, quindi è fondamentale un suo trattamento, che si è osservato essere associato a una riduzione della produzione di citochine pro-infiammatorie nei pazienti con Sm recidivante-remittente.
Rosanna Novara Topino
(quinta puntata – continua)
Approfondimenti
LE?PLACCHE
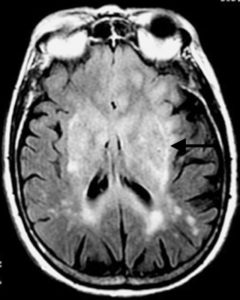 Le placche possono formarsi in ogni punto del Snc, ma sono più frequenti attorno ai ventricoli degli emisferi e del tronco cerebrale, nelle formazioni ottiche, nei peduncoli cerebellari superiori e medi e nel midollo spinale. Al microscopio ottico, le placche rosee si presentano con segni di infiammazione e sono localizzate prevalentemente in zona perivenulare, contengono linfociti T e plasmacellule mentre la mielina si presenta rigonfiata e incapace di assumere la tipica colorazione istologica di Niessel. Inoltre essa risulta frammentata e i suoi frammenti vengono fagocitati dai macrofagi. Le placche grigie sono croniche, in esse la mielina è andata persa e sostituita dalla deposizione di fibre prodotte dalle cellule connettivali, con conseguente cicatrizzazione o sclerosi, che porta allo stiramento e alla frammentazione degli assoni. Finché questi ultimi non sono lesionati, è possibile una riparazione perché la mielina può rigenerarsi. Quando però gli assoni si lesionano, diventano incapaci di trasmettere l’impulso nervoso. Quest’ultimo peraltro rallenta moltissimo già con la perdita del rivestimento di mielina.
Le placche possono formarsi in ogni punto del Snc, ma sono più frequenti attorno ai ventricoli degli emisferi e del tronco cerebrale, nelle formazioni ottiche, nei peduncoli cerebellari superiori e medi e nel midollo spinale. Al microscopio ottico, le placche rosee si presentano con segni di infiammazione e sono localizzate prevalentemente in zona perivenulare, contengono linfociti T e plasmacellule mentre la mielina si presenta rigonfiata e incapace di assumere la tipica colorazione istologica di Niessel. Inoltre essa risulta frammentata e i suoi frammenti vengono fagocitati dai macrofagi. Le placche grigie sono croniche, in esse la mielina è andata persa e sostituita dalla deposizione di fibre prodotte dalle cellule connettivali, con conseguente cicatrizzazione o sclerosi, che porta allo stiramento e alla frammentazione degli assoni. Finché questi ultimi non sono lesionati, è possibile una riparazione perché la mielina può rigenerarsi. Quando però gli assoni si lesionano, diventano incapaci di trasmettere l’impulso nervoso. Quest’ultimo peraltro rallenta moltissimo già con la perdita del rivestimento di mielina.
R.N.T.
LA?DIAGNOSI
Caratteristica della Sm è la sintesi nel liquor cefalorachidiano di IgG (immunoglobuline G, cioè anticorpi) per cui, per porre la diagnosi di Sm, oltre alla valutazione dei segni clinici, alla risonanza magnetica nucleare (Rnm), ai potenziali evocati, si fa ricorso anche a un prelievo di liquor per verificare la presenza di abnormi quantità di IgG endogene.
R.N.T.
Siti web:


 Attualmente non esiste una cura efficace per la Sla. L’unico farmaco approvato dalla Fda (Federal drugs administration statunitense) in grado di rallentarne il decorso per qualche mese è il riluzolo, che in Italia viene somministrato solo a livello ospedaliero.
Attualmente non esiste una cura efficace per la Sla. L’unico farmaco approvato dalla Fda (Federal drugs administration statunitense) in grado di rallentarne il decorso per qualche mese è il riluzolo, che in Italia viene somministrato solo a livello ospedaliero.